









")






Produktinformationen "Hydatid Disease Affecting the Heart and Aorta"
Klinische Vorgeschichte
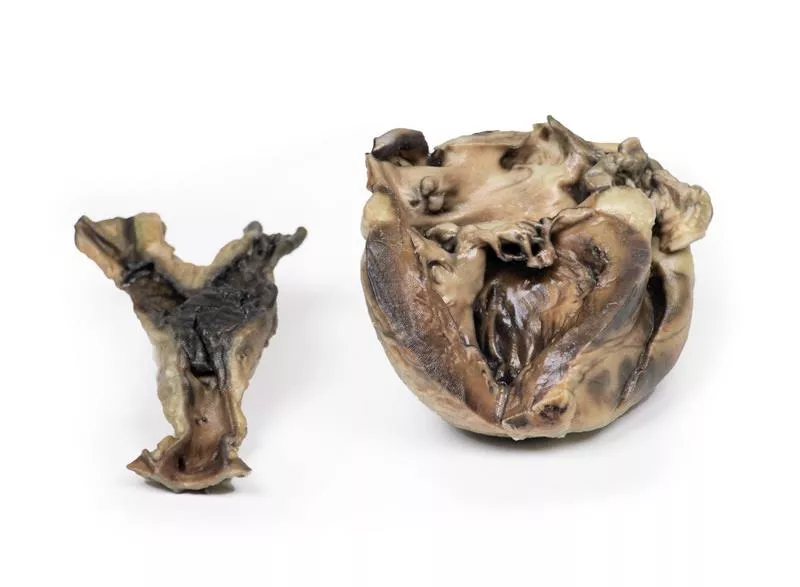
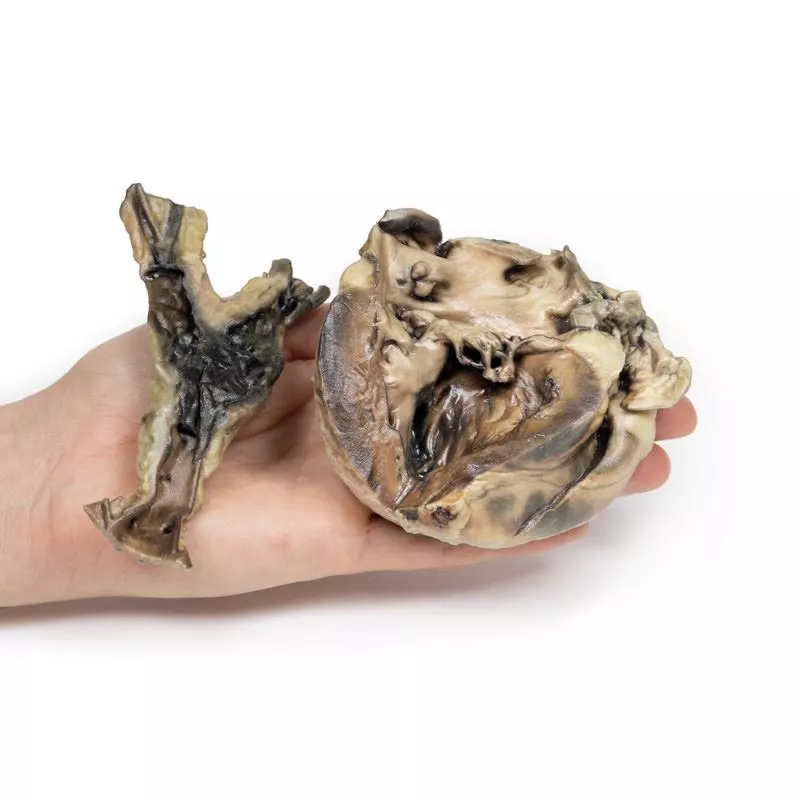
Ein 11-jähriges Mädchen mit einer 18-monatigen Vorgeschichte der Hydatidose hatte in drei Operationen insgesamt 17 Gehirnzysten entfernt bekommen. Weitere Zysten wurden später an Niere, Mesenterium und in der abdominalen Aortenbifurkation entdeckt. Ein Herzröntgen zeigte eine verkalkte Zyste – bei der Entfernung verstarb die Patientin. Im linken Ventrikel fand sich eine tote Hydatidzyste.
Pathologie
Das Herz, geöffnet im Bereich des linken Ventrikels, zeigt deutliche Linksherzhypertrophie und einen ungewöhnlichen Kanal durch den hinteren Segel der Mitralklappe in den linken Vorhof – umgeben von fibrösem Gewebe mit sichtbaren Operationsnähten. Die Bauchaorta an der Iliabifurkation enthält ein großes postmortales Hämatom. Hydatidzysten finden sich in der Aortenwand und im Ventrikel-Vorhof-Kanal, mit typischen Schichten und Tochterzysten sowie Granulomreaktionen.
Weitere Informationen
Die Hydatidose wird durch den Bandwurm Echinococcus granulosus verursacht und über Hundekot übertragen. Menschen infizieren sich durch orale Aufnahme, Zysten bilden sich meist in Leber und Lunge. Die kardiale Beteiligung ist selten (0,5–2?%). Perikardkomplikationen können bei Ruptur auftreten, z.?B. Tamponade.
Ein 11-jähriges Mädchen mit einer 18-monatigen Vorgeschichte der Hydatidose hatte in drei Operationen insgesamt 17 Gehirnzysten entfernt bekommen. Weitere Zysten wurden später an Niere, Mesenterium und in der abdominalen Aortenbifurkation entdeckt. Ein Herzröntgen zeigte eine verkalkte Zyste – bei der Entfernung verstarb die Patientin. Im linken Ventrikel fand sich eine tote Hydatidzyste.
Pathologie
Das Herz, geöffnet im Bereich des linken Ventrikels, zeigt deutliche Linksherzhypertrophie und einen ungewöhnlichen Kanal durch den hinteren Segel der Mitralklappe in den linken Vorhof – umgeben von fibrösem Gewebe mit sichtbaren Operationsnähten. Die Bauchaorta an der Iliabifurkation enthält ein großes postmortales Hämatom. Hydatidzysten finden sich in der Aortenwand und im Ventrikel-Vorhof-Kanal, mit typischen Schichten und Tochterzysten sowie Granulomreaktionen.
Weitere Informationen
Die Hydatidose wird durch den Bandwurm Echinococcus granulosus verursacht und über Hundekot übertragen. Menschen infizieren sich durch orale Aufnahme, Zysten bilden sich meist in Leber und Lunge. Die kardiale Beteiligung ist selten (0,5–2?%). Perikardkomplikationen können bei Ruptur auftreten, z.?B. Tamponade.
Anmelden
Erler-Zimmer
Erler-Zimmer Medical GmbH
Hauptstrasse 27
77886 Lauf
Germany
info@erler-zimmer.de
Achtung! Medizinisches Ausbildungsmaterial, kein Spielzeug. Nicht geeignet für Personen unter 14 Jahren.
Attention! Medical training material, not a toy. Not suitable for persons under 14 years of age.
Andere Kunden kauften auch

Congenital Pulmonary Stenosis
Klinische Vorgeschichte Bei diesem männlichen Kind wurde direkt nach der Geburt ein Herzgeräusch festgestellt. Bis zum 10. Lebensmonat blieb es jedoch unauffällig. Zwei Wochen vor der Krankenhauseinweisung wurde das Kind zunehmend schlapp, bekam leichten Husten und nahm schnell an Gewicht zu. Zehn Tage vor der Aufnahme kam es zu Schwellungen an Gesicht, Händen und Füßen. Bei der Untersuchung war das Kind aufgedunsen, der Blutdruck betrug 90/59 mmHg. Ein feines Schwirren war über dem gesamten Herzbereich zu spüren, das systolische Geräusch war am lautesten im Bereich der Pulmonalklappe. Es bestanden Ödeme an Gesicht und Beinen. Trotz Behandlung kam es zu keiner Besserung, das Kind verstarb an Herzinsuffizienz.Pathologie Das Präparat zeigt das Herz des Kindes in linker Ansicht. Die Pulmonalarterie ist eröffnet und zeigt eine verdickte, konische Pulmonalklappe mit nur 2 mm großer Öffnung. Es besteht eine deutliche poststenotische Erweiterung der Arterie. Das Herz ist auf der rechten Seite stark vergrößert mit einer ausgeprägten Erweiterung des rechten Vorhofs sowie einer rechtsventrikulären Hypertrophie. Es handelt sich um eine isolierte pulmonale Klappenstenose. Weitere Informationen Etwa 7?% aller angeborenen Herzfehler entfallen auf die Pulmonalstenose. Sie kann isoliert oder Teil komplexerer Fehlbildungen wie der Fallot-Tetralogie sein. Der Schweregrad reicht von mild bis schwer. Während milde Formen oft unbemerkt bleiben, können mittelschwere Formen rasch fortschreiten. Eine schwere Stenose führt zu erhöhtem Druck im rechten Herzen, Hypertrophie und letztlich zu Herzinsuffizienz. Weitere Komplikationen sind infektiöse Endokarditis und Herzrhythmusstörungen infolge von Narbenbildung. Die Diagnose erfolgt meist mittels Echokardiografie, weitere Bildgebung kann per MRT oder CT erfolgen. Eine chirurgische Korrektur ist heute gut möglich.

Abdominal Aortic Aneurysm
Klinische Vorgeschichte Ein 70-jähriger Mann mit bekannter leichter gastroösophagealer Refluxkrankheit entwickelte plötzlich starke Oberbauchschmerzen, die bis zur linken Schulter ausstrahlten. Bei der Untersuchung war er unruhig, hyperventilierend, Puls 87/min, Blutdruck 140/90 mmHg. Die Bauchuntersuchung zeigte eine brettharte Abwehr und verminderte Darmgeräusche. Die Notfall-Laparotomie ergab keinen Hinweis auf ein perforiertes Organ, die Bauchspeicheldrüse war unauffällig, jedoch wurde ein nicht rupturiertes Bauchaortenaneurysma festgestellt. Am Folgetag zeigte die Endoskopie ein rupturiertes ösophageales Ulkus, ein Celestin-Schlauch wurde gelegt. Trotz Behandlung kam es zu lokalen Infektionen, Lungenödem und Stauung; der Patient verstarb 19 Tage nach Aufnahme.Pathologie Das Präparat zeigt den unteren Abschnitt der Bauchaorta mit den gemeinsamen und proximalen inneren und äußeren Beckengefäßen. Ein großes 10 x 7 cm großes Aneurysma befindet sich unterhalb der Nierenarterien und reicht bis zur Aortenaufteilung. Die Wand des Aneurysmas ist stark ausgedünnt und teilweise von laminiertem Thrombus bedeckt, was auf einen chronischen Prozess hinweist. Frischer Thrombus ist ebenfalls erkennbar. Es besteht eine Erweiterung der gemeinsamen Beckenarterien und der linken äußeren Beckenschlagader. Im oberen Aortenbereich finden sich multiple fokal ulcerierte atheromatöse Plaques. Eine Ruptur liegt nicht vor. Weitere Informationen Ein Bauchaortenaneurysma (AAA) ist eine örtlich begrenzte Erweiterung der Bauchaorta (Durchmesser >3 cm oder >50% größer als normal). Meistens sind AAAs symptomlos, außer bei Ruptur. Große Aneurysmen können tastbar sein, gelegentlich treten Bauch-, Rücken- oder Beinschmerzen auf. Eine Ruptur führt zu plötzlichen Schmerzen, niedrigem Blutdruck, Bewusstlosigkeit und häufig zum Tod. AAAs treten überwiegend bei Männern über 50 auf, besonders bei Rauchern, Bluthochdruck oder familiärer Belastung. Genetische Erkrankungen wie das Marfan- oder Ehlers-Danlos-Syndrom erhöhen ebenfalls das Risiko. Rund 85 % der AAAs liegen unterhalb der Nierenarterien und sind damit die häufigste Form des Aortenaneurysmas.

Bicuspid Aortic Valve
Klinische Vorgeschichte Eine 64-jährige Frau klagte über Brustschmerzen seit 5 Monaten, begleitet von Atemnot und Keuchen seit 4 Monaten. Bei der Untersuchung zeigte sie Dyspnoe, exspiratorisches Keuchen, linksseitige Rasselgeräusche und Zeichen eines rechtsseitigen Pleuraergusses. Puls und Blutdruck waren normal. Ein präkordialer systolischer Herzton und ein kräftiger Herzspitzenstoß im 5. linken Zwischenrippenraum wurden festgestellt. Periphere Ödeme lagen nicht vor. Die Patientin verstarb 4 Tage nach Aufnahme.Pathologie Das Herz wurde geöffnet und zeigte eine zweisegelige Aortenklappe mit zwei statt drei Klappensegeln, die nur geringfügig verdickt waren. Die Koronararterien, einschließlich der linken Zirkumflexarterie, waren weit offen. Am Herzen fanden sich dichte perikardiale Verwachsungen und Fibrose, Hinweis auf eine konstringierende Perikarditis. Die Autopsie ergab außerdem Aszites, eine kleine zirrhotische Leber, beidseitige Pleuraergüsse und einen Kollaps der rechten Lunge. Todesursache war Leberzirrhose mit Versagen, möglicherweise durch die Perikarditis bedingt. Die zweisegelige Aortenklappe war ein Zufallsbefund. Weitere Informationen Die zweisegelige Aortenklappe ist eine häufige angeborene Anomalie, die oft erst im höheren Alter Symptome zeigt. Sie begünstigt die Entwicklung einer verkalkten Aortenstenose, meist zwischen dem 50. und 70. Lebensjahr. Die Klappe kann isoliert oder Teil eines Syndroms wie der Fallot-Tetralogie sein. Die Fusion von zwei Klappensegeln führt zu ungleich großen Segeln, was abnorme Bewegungen und Turbulenzen verursacht. Dadurch steigt das Risiko für Aortendilatation, Dissektion und Verkalkung. Im Verlauf kann es zu Aortenstenose oder -insuffizienz kommen, die sich durch Atemnot und verminderte Belastbarkeit bemerkbar machen. Die Diagnose wird durch transthorakale Echokardiografie bestätigt.

Calcified Aortic Valvular Stenosis Bicuspid Aortic Valve
Klinische Vorgeschichte Für dieses Präparat liegt keine klinische Vorgeschichte vor.PathologieDas Präparat ist ein etwa 1,5 cm dicker horizontaler Schnitt durch die Ebene des linken Vorhofs. Sichtbar sind die glatte Innenfläche des Vorhofs, das linke Herzohr sowie ein Teil der linken Herzkammer. Auf der oberen Seite erkennt man den Truncus pulmonalis mit einem Teil der pulmonalen Trikuspidalklappe sowie die Aorta mit einer abnormen bikuspiden Aortenklappe. An dieser Klappe finden sich verkalkte Verdickungen an den gegenüberliegenden Rändern. Zudem liegt eine Verkalkung an einer Pulmonalklappentasche vor. Weitere InformationenEine bikuspide Aortenklappe ist der häufigste angeborene Herzfehler und bleibt oft bis ins Erwachsenenalter unentdeckt. Sie kann zu Aortenklappenstenose führen, deren Schweregrad variieren kann. Typische Symptome sind Herzgeräusche, Brustschmerzen, Kurzatmigkeit, Palpitationen sowie Anzeichen einer Herzinsuffizienz wie Müdigkeit und Ödeme. Die Verengung zwingt den linken Ventrikel zu erhöhter Arbeit, was zu Wandverdickung, Erweiterung und letztlich zur Herzschwäche führen kann. Eine Verkalkung der Klappentaschen trägt zusätzlich zur Versteifung und Verengung bei.

Rheumatic endocarditis
Klinische Vorgeschichte Eine 52-jährige Frau klagte über zunehmende Atemnot. Ihre Vorgeschichte umfasste Fieber mit wandernden Gelenkschmerzen in der Kindheit nach einem Halsinfekt. Bei der Untersuchung war sie zyanotisch, mit Vorhofflimmern, erhöhtem Jugularvenendruck, einem pan-systolischen Herzgeräusch am Apex, Hepatomegalie und Ödemen. Sie erhielt Digoxin, Furosemid und Penicillin, verstarb jedoch nach einem Herzstillstand.PathologieDas eröffnete Herz zeigt den linken Vorhof und die Kammer. Die Mitralklappe ist verdickt. Die Wand des linken Vorhofs enthält Blut und Fibrin, und im linken Vorhofohr befindet sich ein Thrombus durch Vorhofflimmern. Ein Wandthrombus und unregelmäßige Endokardverdickungen – die sogenannten MacCallum-Plaques – sind charakteristisch. Weitere InformationenDie Befunde sprechen stark für eine rheumatische Fiebererkrankung, eine autoimmune Folge einer unbehandelten Streptokokkeninfektion. Sie betrifft oft Herz, Gelenke, Haut und Gehirn. Wiederholte Herzbeteiligungen führen zur chronischen rheumatischen Herzkrankheit, meist mit Mitralklappenverengung durch Fibrose. Kennzeichnend sind Aschoff-Knoten. Mit der Zeit dehnt sich der linke Vorhof aus, was zu Vorhofflimmern und Wandthromben führt und schließlich eine schwere Herzinsuffizienz zur Folge haben kann.

Tetralogy of Fallot
Klinische VorgeschichteEin 21 Monate alter Junge wurde mit einer 2–3-monatigen Geschichte von Erschöpfung und Belastungsdyspnoe aufgenommen. Es kam zu mehreren kurzen Episoden akuter Atemnot. Bei der Untersuchung zeigte sich zentrale Zyanose, Klubfinger und ein raues systolisches Herzgeräusch links parasternal. Die Katheteruntersuchung ergab eine Tetralogie von Fallot mit schwerem Lungenödem. Eine Willis-Potts-Anastomose wurde durchgeführt. Der Junge verstarb jedoch 12 Stunden nach der Operation infolge akuter Dyspnoe und Konsolidierung des linken Lungenlappens.PathologieVon vorn betrachtet zeigt das Herz eine Hypertrophie des rechten Ventrikels sowie eine Verengung des Ausflusstrakts zur Pulmonalklappe. Die pulmonale Klappe ist bikuspid und verengt, darunter liegt eine Endokardfibrose. Die Aorta liegt über einem großen Ventrikelseptumdefekt (VSD) und kommuniziert mit dem rechten Ventrikel. Eine Sonde kann vom verengten Truncus pulmonalis über die dilatierte linke Pulmonalarterie durch die Anastomose in die Aorta descendens geführt werden. Rückseitig zeigen sich zwei Atriumseptumdefekte (ASD). Die Wand des linken Ventrikels ist dünner als die des rechten. Weitere InformationenDie Tetralogie von Fallot besteht aus vier Hauptmerkmalen: 1. Ventrikelseptumdefekt, 2. Überreitende Aorta, 3. pulmonale Stenose und 4. rechtsventrikuläre Hypertrophie. Die Zyanose tritt meist früh auf und hängt vom Ausmaß der Obstruktion im Ausflusstrakt ab. Unbehandelte Patienten können unter Umständen bis ins Erwachsenenalter überleben, jedoch ist heute eine chirurgische Korrektur üblich. Zusätzliche Herzfehler wie ASD können vorkommen. Mögliche genetische Ursachen sind Trisomie 21 oder das DiGeorge-Syndrom.

Hypertrophic Subaortic stenosis
Klinische VorgeschichteEin 42-jähriger amerikanischer Tourist wurde tot in seinem Hotelzimmer aufgefunden. Die Todesursache wurde durch eine gerichtliche Obduktion untersucht.PathologieDie Herzpräparation zeigt einen längsgerichteten Schnitt durch beide Ventrikel und das interventrikuläre Septum. Auffällig ist die massive Verdickung des Septums sowie eine Hypertrophie des linken Ventrikels. Die Aorten- und Mitralklappen erscheinen unauffällig. Das verdickte Septum verengt das Lumen des linken Ventrikels.DiagnoseIdiopathische hypertrophe subaortale Stenose, auch bekannt als hypertrophe Kardiomyopathie. Weitere InformationenDie subaortale Stenose ist meist erworben und geht auf eine Fehlbildung im Bereich des Ausflusstrakts des linken Ventrikels (LVOT) zurück. Dadurch entsteht ein turbulenter Blutfluss, der Fibrosierung und Verdickung verursacht. Das Herz muss gegen einen erhöhten Ausflussswiderstand arbeiten, was zur Hypertrophie führt. Milde Verläufe bleiben häufig symptomlos, während fortgeschrittene Formen mit Belastungsdyspnoe oder Synkopen einhergehen können. Ein systolisches Herzgeräusch kann hinweisend sein; die Diagnosesicherung erfolgt per Echokardiographie. Bei schwerer Ausprägung ist die operative Entfernung der Obstruktion angezeigt.

Right Ventricular Hypertrophy
Klinische Vorgeschichte Eine 56-jährige Frau mit Emphysem berichtete über eine zweijährige Zunahme von Atemnot bei Belastung und wiederkehrende Bronchitisanfälle. Bei der Untersuchung betrugen Blutdruck 160/90 mmHg, Puls 96/min, und der Jugularvenendruck war um 6 cm erhöht. Der Herzspitzenstoß war nicht tastbar, beidseitig hörte man Rasselgeräusche, und es bestanden periphere eindrückbare Ödeme. Das EKG zeigte ein Muster einer Rechtsherzbelastung, und die arterielle Blutgasanalyse ergab eine respiratorische Azidose. Trotz Behandlung verschlechterte sich der Zustand stetig bis zum Tod.Pathologie Das Herz von vorne betrachtet zeigte eine stark vergrößerte und hypertrophierte rechte Herzkammer. Ansonsten war das Herz normal. Dies ist ein Beispiel für Rechtsherzhypertrophie (RVH) bei einer Patientin mit Emphysem. Weitere Informationen RVH entsteht meist durch chronische Lungenerkrankungen oder strukturelle Herzfehler. Eine Hauptursache ist die pulmonale Hypertonie (PH), die den Druck in der Pulmonalarterie erhöht und das rechte Herz zur Kompensation durch Hypertrophie zwingt. PH tritt weltweit bei etwa 4 pro Million Menschen auf, wobei in ca. 30 % der Fälle eine RVH vorliegt. Häufige Ursachen für PH sind COPD, Lungenembolie und andere restriktive Lungenerkrankungen. RVH kann auch durch Herzfehler wie Trikuspidalinsuffizienz, Fallot-Tetralogie, Ventrikelseptumdefekte, Pulmonalklappenstenose und Vorhofseptumdefekte verursacht werden. Außerdem steht RVH im Zusammenhang mit Bauchfettleibigkeit und erhöhtem systolischem Blutdruck.

Atrial septal defect
Klinische Vorgeschichte Ein 10-jähriges Mädchen mit bekanntem angeborenem Herzfehler wurde wegen neu aufgetretener Zyanose und Herzinsuffizienz zur Operation aufgenommen. Sie war atemlos, Blutdruck 105/60 mmHg, Puls 140/min. Bei Untersuchung fand man ein lautes Herzgeräusch im 4. linken Zwischenrippenraum nahe dem Sternum, erhöhten Jugularvenendruck und beidseitige basale Rasselgeräusche ohne periphere Ödeme. Der Defekt wurde operativ versorgt, jedoch verstarb das Kind nach einer plötzlichen, ungeklärten postoperativen Verschlechterung.Pathologie Das Herz von links betrachtet zeigte einen großen, 3,5 cm ovalen Defekt im Vorhofseptum mit nur einem kleinen sichelförmigen Rest. Der linke Ventrikel war klein, der rechte hypertrophiert, erkennbar an einer verdickten postero-lateralen Wand. Die Pulmonalarterie war stark erweitert, darüber lag der Aortenbogen, und die linke Vorhofohrmuschel war nahe gelegen. Weitere Informationen Vorhofseptumdefekt (VSD) ist meist im frühen Leben symptomfrei, auch wenn er groß ist. Symptome treten später auf, wenn sich der links-rechts-Shunt durch zunehmende Rechtsherzhypertrophie und pulmonale Hypertonie umkehrt. Dies führt zu einem rechts-links-Shunt mit Zyanose, Atemnot und schließlich Herzinsuffizienz. Formen des VSD sind: - Secundum: Häufigste Form, Mitte des Vorhofseptums. - Primum: Unterer Vorhofseptumabschnitt, oft mit anderen Herzfehlern. - Sinus venosus: Selten, oberer Vorhofseptum, häufig mit weiteren Anomalien. - Coronary sinus: Selten, fehlende Wand zwischen Sinus coronarius und linkem Vorhof.Die Ursachen sind unklar. Manche Herzfehler sind familiär oder mit genetischen Erkrankungen wie Trisomie 21 verbunden. Risiken während der Schwangerschaft sind Infektionen (z.B. Röteln), Drogen- oder Alkoholmissbrauch, toxische Einflüsse und mütterliche Krankheiten wie Diabetes oder Lupus.

Stetige Innovationskraft

Soziale Verantwortung

Gelebte Kundenorientierung

Verständnis für Qualität

Nachhaltiges Handeln

Zertifizierung ISO 9001